
Tất cả nội dung của iLive đều được xem xét về mặt y tế hoặc được kiểm tra thực tế để đảm bảo độ chính xác thực tế nhất có thể.
Chúng tôi có các hướng dẫn tìm nguồn cung ứng nghiêm ngặt và chỉ liên kết đến các trang web truyền thông có uy tín, các tổ chức nghiên cứu học thuật và, bất cứ khi nào có thể, các nghiên cứu đã được xem xét về mặt y tế. Lưu ý rằng các số trong ngoặc đơn ([1], [2], v.v.) là các liên kết có thể nhấp vào các nghiên cứu này.
Nếu bạn cảm thấy rằng bất kỳ nội dung nào của chúng tôi không chính xác, lỗi thời hoặc có thể nghi ngờ, vui lòng chọn nội dung đó và nhấn Ctrl + Enter.
Lissencephaly của não
Chuyên gia y tế của bài báo

Đánh giá lần cuối: 12.07.2025
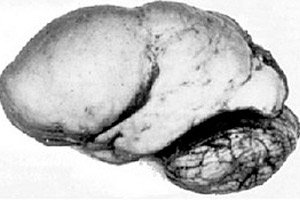
Trong số các bệnh lý não hữu cơ, một dị tật bẩm sinh về sự phát triển của não như bệnh lissencephaly nổi bật, bản chất của nó nằm ở bề mặt gần như nhẵn của vỏ não ở các bán cầu não - với số lượng nếp gấp và rãnh không đủ. [ 1 ]
Trong trường hợp hoàn toàn không có các nếp gấp, agyria được định nghĩa, và sự hiện diện của một số nếp gấp phẳng rộng được gọi là pachygyria. Những khiếm khuyết này, giống như một số biến dạng giảm thiểu khác của não, có mã Q04.3 trong ICD-10.
Dịch tễ học
Theo thống kê về bệnh hiếm gặp, cứ 100 nghìn trẻ sơ sinh thì có 1-1,2 trường hợp mắc bệnh lissencephaly. [ 2 ], [ 3 ]
Theo một số dữ liệu, có tới 25-30% trường hợp lissencephaly cổ điển được quan sát thấy ở trẻ em mắc hội chứng Miller-Dieker; đột biến điểm và mất đoạn gen LIS1 và DCX được phát hiện ở gần 85% bệnh nhân. [ 4 ]
Các nghiên cứu di truyền của 17 gen liên quan đến chứng lissencephaly cho thấy đột biến hoặc mất gen LIS1 chiếm 40% bệnh nhân và 23% liên quan đến đột biến DCX, tiếp theo là TUBA1A (5%) và DYNC1H1 (3%).[ 5 ]
Nguyên nhân não phẳng
Tất cả các nguyên nhân đã biết của sự hình thành vỏ não (cortex cerebri) hầu như hoặc hoàn toàn không có nếp gấp và rãnh, làm tăng "khu vực làm việc" của não người và đảm bảo "năng suất" của hệ thần kinh trung ương, đều liên quan đến sự rối loạn trong quá trình phát triển quanh sinh của nó. Nghĩa là, lissencephaly phát triển ở thai nhi. [ 6 ]
Việc không hình thành được các lớp vỏ não của thai nhi trong tình trạng não không phát triển là kết quả của sự di chuyển bất thường của các tế bào thần kinh hình thành nên não hoặc quá trình này kết thúc sớm.
Quá trình này, vốn cần thiết cho quá trình sinh mô não vỏ não, diễn ra trong nhiều giai đoạn từ tuần thứ 7 đến tuần thứ 18 của thai kỳ. Và, do tính nhạy cảm tăng lên đối với các đột biến gen, cũng như nhiều tác động vật lý, hóa học và sinh học tiêu cực khác nhau, bất kỳ sự sai lệch nào so với chuẩn mực đều có thể dẫn đến định vị không chính xác các tế bào thần kinh với khả năng hình thành lớp chất xám dày lên của vỏ não mà không có cấu trúc đặc trưng. [ 7 ]
Trong một số trường hợp, tình trạng não im lìm ở trẻ em có liên quan đến hội chứng Miller-Dieker, Walker-Warburg hoặc Norman-Roberts.
Đọc thêm – Các khiếm khuyết phát triển của não
Các yếu tố rủi ro
Ngoài các đột biến ở một số gen, các yếu tố nguy cơ sinh ra một đứa trẻ mắc khuyết tật nghiêm trọng như vậy bao gồm tình trạng thiếu oxy (thiếu oxy) của thai nhi; cung cấp máu cho não không đủ (thiếu máu cục bộ); tai biến mạch máu não cấp tính dưới dạng đột quỵ quanh sinh; bệnh lý nhau thai; nhiễm trùng do vi-rút ở phụ nữ mang thai (bao gồm cả TORCH); [ 8 ] các vấn đề về chuyển hóa chung và chức năng tuyến giáp; hút thuốc, rượu, chất hướng thần và chất gây nghiện; sử dụng một số loại thuốc; tăng mức độ bức xạ. [ 9 ]
Sinh bệnh học
Không phải tất cả các trường hợp lissencephaly đều có nguyên nhân gây bệnh do bất thường nhiễm sắc thể và đột biến gen. Nhưng một số gen được biết là mã hóa protein đóng vai trò quan trọng trong chuyển động chính xác của tế bào thần kinh nguyên bào và tế bào thần kinh dọc theo các tế bào thần kinh đệm hướng tâm - để hình thành vỏ não. Và đột biến của các gen này dẫn đến bệnh lý này. [ 10 ]
Đặc biệt, đây là những đột biến ngẫu nhiên (không di truyền) của gen LIS1 trên nhiễm sắc thể 17, điều hòa protein vận động tế bào chất của vi ống dynein, cũng như gen DCX trên nhiễm sắc thể X, mã hóa cho protein doublecortin (lissencephalin-X). [ 11 ]. Trong trường hợp đầu tiên, các chuyên gia định nghĩa lissencephaly cổ điển (loại I), trong trường hợp thứ hai – liên kết với X. [ 12 ]
Khi gen FLN1, mã hóa cho phosphoprotein filamin 1, bị xóa, quá trình di chuyển nơ-ron có hướng có thể không bắt đầu, dẫn đến sự vắng mặt hoàn toàn của các nếp gấp (agyria). [ 13 ]
Các đột biến đã được xác định trong gen CDK5, mã hóa một loại enzyme kinase – chất xúc tác cho quá trình trao đổi chất nội bào, điều chỉnh chu kỳ tế bào ở tế bào thần kinh CNS và đảm bảo sự di chuyển bình thường của chúng trong quá trình hình thành cấu trúc não trước khi sinh.
Những thay đổi bất thường trong gen RELN trên nhiễm sắc thể số 7 gây ra các khiếm khuyết hồi vỏ não trong hội chứng Norman-Roberts dẫn đến tình trạng thiếu hụt glycoprotein ngoại bào reelin, một loại glycoprotein cần thiết để điều chỉnh quá trình di chuyển và định vị của các tế bào gốc thần kinh trong quá trình phát triển vỏ não.[ 14 ], [ 15 ], [ 16 ]
Gen ARX mã hóa protein hộp không phải aristalens, một yếu tố phiên mã đóng vai trò quan trọng trong não trước và các mô khác.[ 17 ] Trẻ em bị đột biến ARX có các triệu chứng khác, chẳng hạn như thiếu một số phần của não (thiếu thể chai), cơ quan sinh dục bất thường và động kinh nặng.[ 18 ],[ 19 ]
Một số gen đã được liên kết với lissencephaly. Các gen này bao gồm VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A và CDK5.[ 20 ]
Cytomegalovirus (CMV) có liên quan đến sự phát triển của chứng lissencephaly do giảm lượng máu cung cấp cho não của thai nhi. Mức độ nghiêm trọng của nhiễm CMV phụ thuộc vào tuổi thai. Nhiễm trùng sớm có nhiều khả năng gây ra chứng lissencephaly vì sự di chuyển của tế bào thần kinh xảy ra sớm trong thai kỳ.[ 21 ]
Ngoài ra, cơ chế xảy ra dị tật này bao gồm sự ngừng di chuyển không hoàn toàn hoặc sau đó của các tế bào thần kinh từ vùng sinh sản quanh não thất đến vỏ não. Và trong những trường hợp như vậy, hoặc lissencephaly không hoàn toàn hoặc pachygyria phát triển, trong đó hình thành một số rãnh rộng và nếp gấp (nhưng hầu hết đều không có).
Triệu chứng não phẳng
Những dấu hiệu đầu tiên của bệnh lý này (khi không có các hội chứng đã đề cập trước đó) có thể không xuất hiện ngay sau khi sinh, mà sau một tháng rưỡi đến hai tháng. Và thường gặp nhất là các triệu chứng lâm sàng sau đây của bệnh lissencephaly:
- trương lực cơ thấp, thường kết hợp với liệt cứng;
- co giật và cơn động kinh toàn thể co cứng-co giật (dưới dạng uốn cong);
- chậm phát triển trí tuệ và chậm phát triển nghiêm trọng;
- suy giảm chức năng thần kinh và vận động.
Các vấn đề về nuốt gây khó khăn trong việc cho trẻ bú. [ 22 ]
Mức độ suy giảm thần kinh vận động cao thường biểu hiện dưới dạng tetraplegia – liệt tất cả các chi. Có thể bị biến dạng bàn tay, ngón tay hoặc ngón chân.
Trong hội chứng Norman-Roberts với chứng teo não loại I, các dị tật sọ mặt được quan sát thấy: chứng đầu nhỏ nghiêm trọng, trán thấp và sống mũi rộng nhô ra, mắt cách xa nhau (hyperterlorism), hàm dưới kém phát triển (micrognathia). [ 23 ]
Hội chứng Miller-Dieker cũng có thể được đặc trưng bởi kích thước đầu nhỏ bất thường với trán rộng, cao và mũi ngắn, lõm ở thái dương (trũng hai bên thái dương) và tai thấp, biến dạng.
Hội chứng lissencephaly nghiêm trọng được đặc trưng bởi chứng đầu nhỏ, giảm kích thước nhãn cầu (tật mắt nhỏ), kết hợp với loạn sản võng mạc, não úng thủy tắc nghẽn và không có hoặc giảm sản thể chai.
Các biến chứng và hậu quả
Trong số các biến chứng của dị tật này, các chuyên gia nêu ra tình trạng rối loạn chức năng nuốt (khó nuốt) và trào ngược dạ dày thực quản; động kinh khó chữa (không kiểm soát được); nhiễm trùng đường hô hấp trên thường xuyên; viêm phổi (bao gồm cả tình trạng hít sặc mãn tính).
Trẻ sơ sinh bị chứng lissencephaly có thể có các vấn đề tim bẩm sinh hữu cơ dưới dạng khuyết tật vách ngăn tâm nhĩ hoặc khuyết tật tim phức tạp với chứng tím tái (bốn bệnh Fallot). [ 24 ]
Hậu quả của tình trạng chậm phát triển sau sinh trong hầu hết các trường hợp là tử vong trong vòng 24 tháng sau khi sinh.
Chẩn đoán não phẳng
Chẩn đoán bắt đầu bằng việc khám sức khỏe cho trẻ, tìm hiểu tiền sử bệnh lý của cha mẹ và tiền sử mang thai và sinh nở.
Trong thời kỳ mang thai, có thể cần phải xét nghiệm DNA thai nhi không có tế bào, chọc ối hoặc lấy mẫu nhung mao màng đệm. [ 25 ] Để biết thêm thông tin, hãy xem – Chẩn đoán trước sinh các bệnh bẩm sinh
Chẩn đoán bằng dụng cụ được sử dụng để hình dung cấu trúc não và đánh giá chức năng của chúng:
- chụp cắt lớp vi tính não;
- chụp cộng hưởng từ (MRI) não;
- điện não đồ (EEG). [ 26 ]
Trong thời kỳ mang thai, có thể nghi ngờ tình trạng não teo khi siêu âm thai nhi sau 20-21 tuần do không có rãnh đỉnh chẩm và rãnh xương cựa và bất thường ở khe Sylvian của não.
Chẩn đoán phân biệt
Tiến hành chẩn đoán phân biệt với các hội chứng khác của bệnh lý não bẩm sinh.
Có hơn 20 loại lissencephaly, hầu hết trong số đó thuộc 2 loại chính: lissencephaly cổ điển (loại 1) và lissencephaly đá cuội (loại 2). Mỗi loại có biểu hiện lâm sàng tương tự nhưng đột biến gen khác nhau.[ 27 ]
Kiểm tra não ở bệnh nhân lissencephaly loại I cho thấy vỏ não có bốn lớp thay vì sáu lớp như ở bệnh nhân bình thường, trong khi ở bệnh nhân lissencephaly loại 2, vỏ não không có tổ chức và trông giống như cục u hoặc nốt sần do vỏ não bị dịch chuyển hoàn toàn bởi các cụm tế bào thần kinh vỏ não tách biệt bởi mô trung mô thần kinh đệm. Bệnh nhân cũng có bất thường về cơ và mắt.
- Lissencephaly cổ điển (loại 1):
- LIS1: Lissencephaly đơn độc và hội chứng Miller-Dieker (lissencephaly liên quan đến dị dạng khuôn mặt). [ 28 ]
- LISX1: Đột biến gen DCX. So với chứng lissencephaly do đột biến LIS1, DCX có vỏ não sáu lớp thay vì bốn lớp.
- Lissencephaly đơn độc không có khiếm khuyết di truyền nào khác được biết đến
- Bệnh não phẳng dạng đá cuội (loại 2):
- Hội chứng Walker-Warburg
- Hội chứng Fukuyama
- Bệnh về cơ, mắt và não
- Các loại khác không thể được xếp vào một trong hai nhóm trên:
- LIS2: Hội chứng Norman-Roberts, tương tự như hội chứng lissencephaly loại I hoặc hội chứng Miller-Dieker, nhưng không mất nhiễm sắc thể 17.
- LIS3
- LISX2
Microlissencephaly: Đây là sự kết hợp của việc không có nếp gấp vỏ não bình thường và đầu nhỏ bất thường. Trẻ em bị lissencephaly bình thường có đầu kích thước bình thường khi sinh ra. Trẻ em có kích thước đầu nhỏ khi sinh ra thường được chẩn đoán mắc microlissencephaly.
Điều quan trọng nữa là phải phân biệt giữa tình trạng lissencephaly và polymicrogyria, đây là những khiếm khuyết phát triển não khác nhau.
Ai liên lạc?
Điều trị não phẳng
Lissencephaly là một khiếm khuyết hữu cơ không thể chữa khỏi, do đó chỉ có thể điều trị hỗ trợ và triệu chứng. [ 29 ]
Trước hết, đây là việc sử dụng thuốc chống co giật và thuốc chống động kinh, cũng như việc đặt ống thông dạ dày vào dạ dày (nếu trẻ không thể tự nuốt). Xoa bóp là hữu ích.
Trong trường hợp não úng thủy nghiêm trọng, dịch não tủy sẽ được lấy ra.
Phòng ngừa
Các chuyên gia khuyến cáo rằng các bậc cha mẹ tương lai nên tìm kiếm tư vấn di truyền và phụ nữ mang thai nên đăng ký với bác sĩ sản phụ khoa kịp thời và trải qua tất cả các cuộc kiểm tra theo lịch trình.
Dự báo
Đối với trẻ em bị lissencephaly, tiên lượng phụ thuộc vào mức độ của bệnh, nhưng thường thì sự phát triển về mặt tinh thần của trẻ không vượt quá mức bốn đến năm tháng. Và tất cả trẻ em được chẩn đoán mắc bệnh này đều mắc chứng rối loạn tâm thần vận động nghiêm trọng và động kinh khó điều trị. [ 30 ]
Theo NINDS (Viện Quốc gia về Rối loạn Thần kinh và Đột quỵ Hoa Kỳ), tuổi thọ tối đa của những người mắc chứng lissencephaly là khoảng 10 năm.