
Tất cả nội dung của iLive đều được xem xét về mặt y tế hoặc được kiểm tra thực tế để đảm bảo độ chính xác thực tế nhất có thể.
Chúng tôi có các hướng dẫn tìm nguồn cung ứng nghiêm ngặt và chỉ liên kết đến các trang web truyền thông có uy tín, các tổ chức nghiên cứu học thuật và, bất cứ khi nào có thể, các nghiên cứu đã được xem xét về mặt y tế. Lưu ý rằng các số trong ngoặc đơn ([1], [2], v.v.) là các liên kết có thể nhấp vào các nghiên cứu này.
Nếu bạn cảm thấy rằng bất kỳ nội dung nào của chúng tôi không chính xác, lỗi thời hoặc có thể nghi ngờ, vui lòng chọn nội dung đó và nhấn Ctrl + Enter.
Bệnh Charcot-Marie-Tooth.
Chuyên gia y tế của bài báo

Đánh giá lần cuối: 12.07.2025
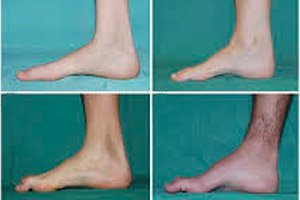
Teo cơ mác, hội chứng hoặc bệnh Charcot-Marie-Tooth là một nhóm bệnh di truyền mãn tính gây tổn thương các dây thần kinh ngoại biên.
Theo ICD-10, trong phần về các bệnh của hệ thần kinh, mã bệnh này là G60.0 (bệnh thần kinh vận động và cảm giác di truyền). Bệnh này cũng được đưa vào danh sách các bệnh mồ côi.
Dịch tễ học
Theo số liệu thống kê lâm sàng, tỷ lệ mắc tất cả các loại bệnh Charcot-Marie-Tooth trên 100 nghìn dân là 19 trường hợp (theo các nguồn khác, một trường hợp trên 2,5-10 nghìn dân).
CMT loại 1 chiếm khoảng hai phần ba số ca bệnh (một ca trên 5.000 đến 7.000 dân số) và gần 70% trong số đó có liên quan đến sự nhân đôi của gen PMP22. Hơn 1,2 triệu người trên toàn thế giới mắc phải loại bệnh này.
Tỷ lệ mắc CMT loại 4 ước tính ở mức 1-5 trường hợp trên 10.000 trẻ em. [ 1 ]
Nguyên nhân Bệnh Charcot-Marie-Tooth
Theo phân loại hội chứng đa dây thần kinh, teo cơ mác (xương mác), teo cơ thần kinh Charcot-Marie-Tooth hoặc bệnh Charcot-Marie-Tooth (viết tắt là CMT) đề cập đến các bệnh lý đa dây thần kinh vận động-cảm giác được xác định về mặt di truyền. [ 2 ]
Nghĩa là, nguyên nhân gây ra bệnh là đột biến gen. Và tùy thuộc vào bản chất của các sai lệch di truyền, các loại hoặc dạng chính của hội chứng này được phân biệt: mất myelin và sợi trục. Nhóm đầu tiên bao gồm bệnh Charcot-Marie-Tooth loại 1 (CMT1), xảy ra do sự nhân đôi của gen PMP22 trên nhiễm sắc thể 17, mã hóa cho protein myelin ngoại vi xuyên màng 22. Kết quả là, mất myelin từng đoạn của bao sợi trục (các quá trình tế bào thần kinh) và giảm tốc độ dẫn truyền tín hiệu thần kinh. Ngoài ra, đột biến có thể xảy ra ở một số gen khác.
Dạng trục là bệnh Charcot-Marie-Tooth loại 2 (CMT2), ảnh hưởng đến chính các trục và liên quan đến những thay đổi bệnh lý ở gen MFN2 tại locus 1p36.22, mã hóa protein màng mitofusin-2, cần thiết cho sự hợp nhất ty thể và hình thành mạng lưới ty thể chức năng bên trong tế bào thần kinh ngoại biên. Có hơn một chục phân nhóm của CMT2 (với các đột biến ở các gen cụ thể).
Cần lưu ý rằng hiện nay đã xác định được hơn một trăm gen có tổn thương, được truyền qua di truyền, gây ra nhiều phân nhóm bệnh Charcot-Marie-Tooth. Ví dụ, đột biến ở gen RAB7 dẫn đến CMT loại 2B; biến đổi gen SH3TC2 (mã hóa một trong các protein màng của tế bào Schwann) gây ra CMT loại 4C, biểu hiện ở trẻ em và đặc trưng bởi tình trạng mất myelin ở các tế bào thần kinh vận động và cảm giác (có một tá rưỡi dạng loại 4 của bệnh này).
Một loại CMT hiếm gặp loại 3 (gọi là hội chứng Dejerine-Sottas) bắt đầu phát triển ở thời thơ ấu và do đột biến ở PMP22, MPZ, EGR2 và các gen khác gây ra.
Khi CMT loại 5 xảy ra ở độ tuổi từ 5-12, không chỉ có bệnh lý thần kinh vận động (dưới dạng liệt cứng hai chi dưới) được quan sát thấy mà còn có tổn thương ở dây thần kinh thị giác và thính giác.
Yếu cơ và teo cơ (kèm theo mất thị lực) cũng như các vấn đề về thăng bằng là đặc trưng của CMT loại 6. Và ở bệnh Charcot-Marie-Tooth loại 7 không chỉ có bệnh lý thần kinh vận động-cảm giác mà còn có bệnh lý võng mạc dưới dạng viêm võng mạc sắc tố.
Phổ biến hơn ở nam giới, bệnh CMT liên kết X hoặc bệnh Charcot-Marie-Tooth với chứng liệt tứ chi (yếu khả năng vận động của cả tay và chân) là loại mất myelin và được cho là kết quả của đột biến ở gen GJB1 trên cánh tay dài của nhiễm sắc thể X, mã hóa connexin 32, một protein xuyên màng của tế bào Schwann và tế bào ít nhánh điều chỉnh quá trình truyền tín hiệu thần kinh. [ 3 ]
Các yếu tố rủi ro
Yếu tố nguy cơ chính của CMT là tiền sử gia đình mắc bệnh, tức là có người thân gần.
Theo các nhà di truyền học, nếu cả cha và mẹ đều là người mang gen lặn trên nhiễm sắc thể thường của bệnh Charcot-Marie-Tooth, nguy cơ sinh ra đứa con mắc bệnh này là 25%. Và nguy cơ đứa trẻ là người mang gen này (nhưng không có bất kỳ triệu chứng nào) ước tính là 50%.
Trong trường hợp di truyền liên kết với nhiễm sắc thể X (khi gen đột biến nằm trên nhiễm sắc thể X của người phụ nữ), có 50% nguy cơ người mẹ sẽ truyền gen cho con trai, người sẽ phát triển CMT. Bệnh có thể không xảy ra khi một đứa trẻ gái được sinh ra, nhưng con trai của người con gái (cháu) có thể thừa hưởng gen khiếm khuyết và bệnh sẽ phát triển.
Sinh bệnh học
Trong bất kỳ loại bệnh Charcot-Marie-Tooth nào, cơ chế sinh bệnh của nó đều do bất thường di truyền của các dây thần kinh ngoại biên: vận động (chuyển động) và cảm giác (nhạy cảm).
Nếu loại CMT là mất myelin, thì sự phá hủy hoặc khiếm khuyết của bao myelin bảo vệ sợi trục của dây thần kinh ngoại biên sẽ dẫn đến sự chậm lại trong việc truyền xung thần kinh trong hệ thần kinh ngoại biên - giữa não, cơ và các cơ quan cảm giác.
Ở loại bệnh liên quan đến sợi trục, bản thân các sợi trục bị ảnh hưởng, ảnh hưởng tiêu cực đến cường độ tín hiệu thần kinh, không đủ để kích thích đầy đủ các cơ và các cơ quan cảm giác.
Đọc thêm:
Hội chứng Charcot-Marie-Tooth lây truyền như thế nào? Các gen khiếm khuyết có thể được di truyền theo kiểu trội trên nhiễm sắc thể thường hoặc lặn trên nhiễm sắc thể thường.
Loại phổ biến nhất, di truyền trội trên nhiễm sắc thể thường, xảy ra khi có một bản sao của gen đột biến (do một trong hai cha mẹ mang). Và xác suất truyền CMT cho mỗi đứa trẻ được sinh ra ước tính là 50%. [ 4 ]
Với sự di truyền lặn trên nhiễm sắc thể thường, bệnh cần có hai bản sao của gen khiếm khuyết (một từ mỗi bên cha mẹ không biểu hiện bất kỳ dấu hiệu nào của bệnh) để phát triển.
Trong 40-50% trường hợp, mất myelin di truyền trội trên nhiễm sắc thể thường xảy ra, tức là CMT loại 1; trong 12-26% trường hợp, CMT sợi trục, tức là loại 2. Và trong 10-15% trường hợp, di truyền liên kết với X được quan sát thấy. [ 5 ]
Triệu chứng Bệnh Charcot-Marie-Tooth
Thông thường, các dấu hiệu đầu tiên của căn bệnh này bắt đầu xuất hiện ở thời thơ ấu và thanh thiếu niên và phát triển dần dần trong suốt cuộc đời, mặc dù hội chứng này có thể tự biểu hiện sau này. Sự kết hợp của các triệu chứng là khác nhau và tốc độ tiến triển của bệnh, cũng như mức độ nghiêm trọng của nó, là không thể dự đoán được.
Các triệu chứng điển hình của giai đoạn đầu bao gồm mệt mỏi toàn thân tăng lên; giảm trương lực (yếu) của các cơ ở bàn chân, mắt cá chân và cẳng chân; thiếu phản xạ. Điều này làm cho việc di chuyển bàn chân trở nên khó khăn và dẫn đến loạn dưỡng (rối loạn dáng đi) dưới dạng chân giơ cao hơn, thường xuyên vấp ngã. Các dấu hiệu của bệnh Charcot-Marie-Tooth ở trẻ nhỏ có thể bao gồm sự vụng về rõ rệt và khó khăn khi đi lại không đặc trưng theo độ tuổi liên quan đến chứng bàn chân rủ hai bên. Các dị tật ở bàn chân cũng là đặc trưng: vòm bàn chân cao (bàn chân rỗng) hoặc bàn chân phẳng nghiêm trọng, ngón chân cong (hình búa).
Trong trường hợp trẻ đi bằng đầu ngón chân trên nền cơ trương lực thấp, bác sĩ thần kinh có thể nghi ngờ trẻ mắc CMT loại 4, trong đó trẻ có thể không thể đi lại khi đến tuổi vị thành niên.
Khi bệnh tiến triển, teo cơ và yếu cơ lan đến các chi trên, khiến việc thực hiện các kỹ năng vận động tinh và thực hiện các nhiệm vụ bình thường của tay trở nên khó khăn. Giảm cảm giác xúc giác và khả năng cảm nhận nhiệt độ nóng lạnh, cũng như tê ở bàn chân và bàn tay, cho thấy tổn thương ở các sợi trục của dây thần kinh cảm giác.
Ở bệnh Charcot-Marie-Tooth loại 3 và 6 biểu hiện ở trẻ em, có thể thấy tình trạng mất điều hòa cảm giác (suy giảm khả năng phối hợp các chuyển động và thăng bằng), co giật và run cơ, tổn thương dây thần kinh mặt, teo dây thần kinh thị giác kèm theo rung giật nhãn cầu và mất thính lực.
Ở giai đoạn sau, có thể có tình trạng run rẩy không kiểm soát được và chuột rút cơ thường xuyên; các vấn đề về vận động có thể dẫn đến phát triển các cơn đau: cơ, khớp, thần kinh.
Các biến chứng và hậu quả
Bệnh Charcot-Marie-Tooth có thể gây ra các biến chứng và hậu quả như sau:
- bong gân và gãy xương thường xuyên hơn;
- co cứng liên quan đến sự co ngắn của các cơ và gân quanh khớp;
- vẹo cột sống (độ cong của cột sống);
- các vấn đề về hô hấp – khi các sợi thần kinh chi phối các cơ hoành bị tổn thương:
- mất khả năng di chuyển độc lập.
Chẩn đoán Bệnh Charcot-Marie-Tooth
Chẩn đoán bao gồm khám lâm sàng, tiền sử bệnh (bao gồm cả tiền sử gia đình), khám thần kinh và toàn thân.
Các xét nghiệm được thực hiện để kiểm tra phạm vi chuyển động, độ nhạy và phản xạ gân. Có thể đánh giá dẫn truyền thần kinh bằng cách sử dụng chẩn đoán bằng dụng cụ – điện cơ đồ hoặc điện thần kinh đồ. Siêu âm hoặc MRI cũng có thể được yêu cầu. [ 6 ]
Xét nghiệm di truyền hoặc DNA để phát hiện các đột biến di truyền phổ biến nhất gây ra CMT trong mẫu máu bị hạn chế vì hiện tại không có xét nghiệm DNA cho tất cả các loại bệnh. Để biết thêm thông tin, hãy xem Xét nghiệm di truyền
Trong một số trường hợp, cần phải sinh thiết dây thần kinh ngoại biên (thường là dây thần kinh sural).
Chẩn đoán phân biệt
Chẩn đoán phân biệt bao gồm các bệnh lý thần kinh ngoại biên khác, bệnh loạn dưỡng cơ Duchenne, hội chứng tủy sống và nhược cơ, bệnh lý thần kinh do đái tháo đường, bệnh lý tủy sống trong bệnh xơ cứng rải rác và teo cơ một bên, hội chứng Guillain-Barré, chấn thương dây thần kinh mác và teo dây thần kinh này (bao gồm cả khi bị chèn ép giữa các đĩa đệm thắt lưng của cột sống), tổn thương tiểu não hoặc đồi thị, cũng như tác dụng phụ của hóa trị liệu (trong quá trình điều trị bằng thuốc chống tế bào như Vincristine hoặc Paclitaxel). [ 7 ]
Ai liên lạc?
Điều trị Bệnh Charcot-Marie-Tooth
Ngày nay, phương pháp điều trị căn bệnh di truyền này bao gồm liệu pháp tập thể dục (nhằm tăng cường và kéo giãn cơ); liệu pháp nghề nghiệp (giúp bệnh nhân bị yếu cơ ở tay); và sử dụng các thiết bị chỉnh hình để hỗ trợ đi bộ. Nếu cần thiết, thuốc giảm đau hoặc thuốc chống co giật sẽ được kê đơn. [ 8 ]
Trong trường hợp bàn chân bẹt nghiêm trọng, có thể tiến hành phẫu thuật cắt xương, và trong trường hợp biến dạng gót chân, có chỉ định phẫu thuật chỉnh hình – cố định khớp. [ 9 ]
Nghiên cứu đang được tiến hành về cả thành phần di truyền của bệnh và phương pháp điều trị. Việc sử dụng tế bào gốc, một số hormone, lecithin hoặc axit ascorbic vẫn chưa mang lại kết quả tích cực.
Nhưng nhờ vào nghiên cứu gần đây, một cái gì đó mới thực sự có thể xuất hiện trong việc điều trị bệnh Charcot-Marie-Tooth trong tương lai gần. Do đó, kể từ năm 2014, công ty Pharnext của Pháp đã phát triển và từ giữa năm 2019, các thử nghiệm lâm sàng đã được tiến hành đối với loại thuốc PXT3003 để điều trị CMT loại 1 ở người lớn, có tác dụng ức chế sự biểu hiện tăng lên của gen PMP22, cải thiện quá trình tạo myelin của dây thần kinh ngoại biên và làm giảm các triệu chứng thần kinh cơ.
Các chuyên gia từ công ty y khoa Sarepta Therapeutics (Hoa Kỳ) đang nghiên cứu tạo ra liệu pháp gen cho bệnh Charcot-Marie-Tooth loại 1. Liệu pháp này sẽ sử dụng một loại virus liên kết adeno (AAV) vô hại thuộc chi Dependovirus với bộ gen DNA mạch đơn tuyến tính, sẽ chuyển gen NTF3 vào cơ thể, mã hóa protein neurotrophin-3 (NT-3) cần thiết cho hoạt động của tế bào thần kinh Schwann.
Helixmith sẽ bắt đầu thử nghiệm lâm sàng liệu pháp gen Engensis (VM202) do Hàn Quốc phát triển vào cuối năm 2020 để điều trị các triệu chứng cơ ở bệnh nhân CMT loại 1. [ 10 ]
Phòng ngừa
Phòng ngừa CMT có thể là tư vấn di truyền cho cha mẹ tương lai, đặc biệt là nếu một trong hai người có tiền sử gia đình mắc bệnh. Tuy nhiên, đã xác định được các trường hợp đột biến gen điểm de novo, tức là không có bệnh trong tiền sử gia đình.
Trong thời kỳ mang thai, khả năng mắc bệnh Charcot-Marie-Tooth ở trẻ sau này có thể được kiểm tra bằng cách sinh thiết nhung mao màng đệm (từ tuần thứ 10 đến tuần thứ 13 của thai kỳ) cũng như xét nghiệm nước ối (từ tuần thứ 15 đến tuần thứ 18).
Dự báo
Nhìn chung, tiên lượng cho các loại bệnh Charcot-Marie-Tooth khác nhau phụ thuộc vào mức độ nghiêm trọng lâm sàng, nhưng trong mọi trường hợp, bệnh đều tiến triển chậm. Nhiều bệnh nhân bị khuyết tật, mặc dù điều này không làm giảm tuổi thọ.